
Unsere Forschung
Wir sind eine bioinformatische Forschungsgruppe, die sich mit der Untersuchung von Viren und Virusinfektionen unter Verwendung sequenzbasierter Ansätze befasst. Ein roter Faden unserer Projekte ist die Nutzung von Rohdaten aus öffentlichen Sequenzierungsarchiven, um in enger Zusammenarbeit mit experimentellen Virolog:innen und Kliniker:innen wissenschaftliche Hypothesen zu entwickeln und zu überprüfen. Wir verwenden Hochleistungsrechner und Deep Learning Ansätze, um die stetig wachsenden Daten- und Metadatenmengen zu analysieren.
Datengestützte Virusentdeckung
Ein Schwerpunkt unserer Forschung ist die Untersuchung der genetischen Vielfalt von Viren und der Virusevolution. Wir nutzen dafür große Mengen öffentlicher Sequenzierungsdaten, die wir nach viralen Genomen, welche als Nebenprodukt der Sequenzierung des Genoms oder Transkriptoms des untersuchten Organismus sequenziert wurden, zu durchsuchen. Ein Hauptziel unserer Forschung ist es, das eukaryotische Virom sowie das gesamte Virom von erkrankten und gesunden Menschen umfassend zu charakterisieren. Darüber hinaus versuchen wir, unbekannte Tierviren zu entdecken, die eng mit pathogenen Humanviren verwandt sind und somit als experimentelle Modelle dienen können. Unser besonderes Interesse gilt den Nidoviren und der Frage, wie sie die größten bekannten RNA-Genome mit einer Länge von bis zu 64 Kilobasen entwickelt haben. Wir koordinieren die taxonomische Klassifizierung von Nidoviren im Rahmen des International Committee on Taxonomy of Viruses (ICTV).
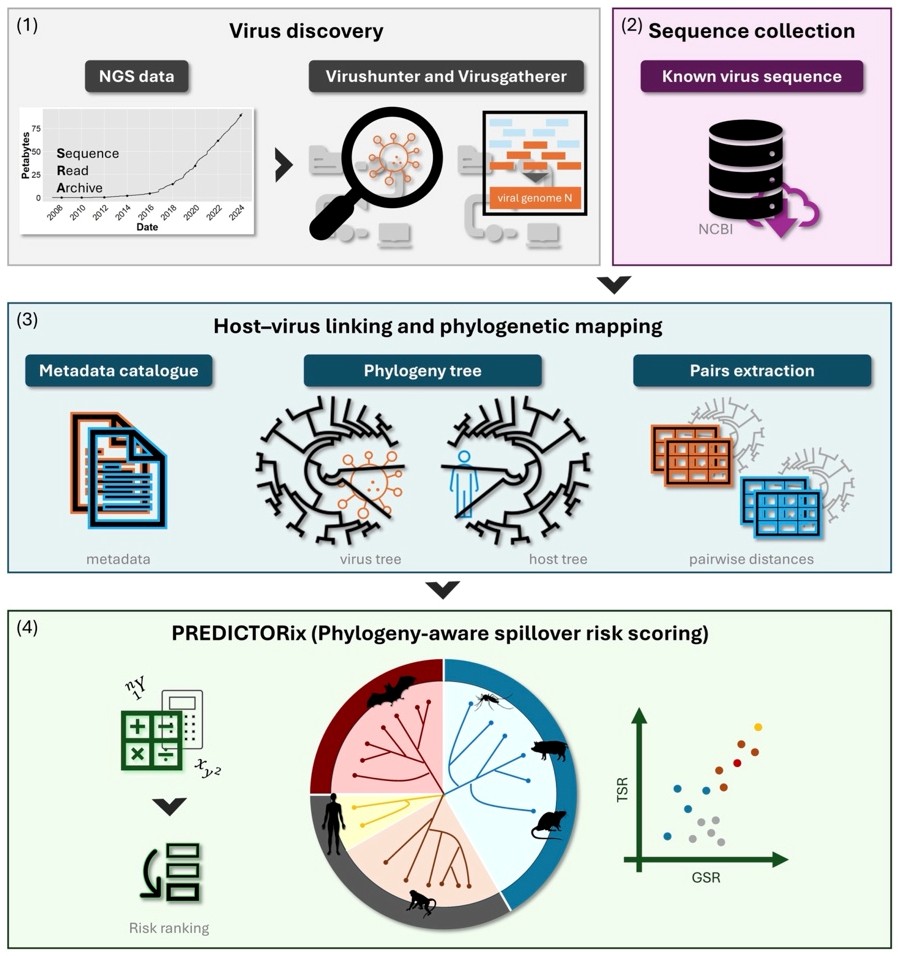
PREDICTORix – ein Ansatz zur Quantifizierung des Risikos zoonotischer Übertragungen basierend auf Virus-Wirt Koevolution
PREDICTORix ist ein quantitativer, phylogeniebasierter Ansatz, um das Risiko zoonotischer Übertragungen über ein breites Spektrum von Virusfamilien hinweg abzuschätzen, ohne dass experimentelle Daten zu Übertragung oder Pathogenität erforderlich sind. Durch die Kombination globaler Sequenzressourcen (einschließlich >150.000 neu entdeckter RNA-Virussequenzen), der expliziten Modellierung evolutionärer Beziehungen zwischen Virus und Wirt sowie zweier Risikometriken (allgemeines und gezieltes Spillover-Risiko) generiert PREDICTORix quantitative Spillover-Werte, die als Leitfaden für Interventionen und mechanistische Folgemaßnahmen dienen können. Ursprünglich als Resilienzmaßnahme gegenüber menschlichen Pandemien entwickelt, lässt sich der Ansatz auf Tiere und andere Wirtsarten ausweiten und hilft dabei, potenzielle Kandidaten für „Pathogen X“ zu antizipieren.
Genetische Determinanten schwerer RSV-Infektionen
In enger Zusammenarbeit mit RESIST-Wissenschaftler:innen und Kliniker:innen der MHH untersuchen wir das komplexe Zusammenspiel genetischer Variationen im menschlichen Genom, um genetische Veränderungen zu identifizieren, die mit dem Verlauf oder der Schwere einer Infektion mit dem humanen Respiratorischen Synzytial-Virus (RSV) bei Kleinkindern assoziiert sind. Wir versuchen, sowohl monogene als auch polygene Faktoren zu identifizieren, die den Schweregrad der Erkrankung beeinflussen. Um diese und andere Fragen zu klären, haben unsere lokalen und internationalen Kooperationspartner die Exome von Kleinkindern mit schwerer RSV-Erkrankung sowie von Kontrollpersonen mit leichter RSV-Infektion sequenziert. Wir wenden Bioinformatik und genetische Assoziationsanalysen an, um wahrscheinliche kausale Varianten in immunbezogenen Genen zu identifizieren, die die Grundlage für experimentelle Folgeuntersuchungen bilden.
Unsere Forschung
Wir sind eine bioinformatische Forschungsgruppe, die sich mit der Untersuchung von Viren und Virusinfektionen unter Verwendung sequenzbasierter Ansätze befasst. Ein roter Faden unserer Projekte ist die Nutzung von Rohdaten aus öffentlichen Sequenzierungsarchiven, um in enger Zusammenarbeit mit experimentellen Virolog:innen und Kliniker:innen wissenschaftliche Hypothesen zu entwickeln und zu überprüfen. Wir verwenden Hochleistungsrechner und Deep Learning Ansätze, um die stetig wachsenden Daten- und Metadatenmengen zu analysieren.
Datengestützte Virusentdeckung
Ein Schwerpunkt unserer Forschung ist die Untersuchung der genetischen Vielfalt von Viren und der Virusevolution. Wir nutzen dafür große Mengen öffentlicher Sequenzierungsdaten, die wir nach viralen Genomen, welche als Nebenprodukt der Sequenzierung des Genoms oder Transkriptoms des untersuchten Organismus sequenziert wurden, zu durchsuchen. Ein Hauptziel unserer Forschung ist es, das eukaryotische Virom sowie das gesamte Virom von erkrankten und gesunden Menschen umfassend zu charakterisieren. Darüber hinaus versuchen wir, unbekannte Tierviren zu entdecken, die eng mit pathogenen Humanviren verwandt sind und somit als experimentelle Modelle dienen können. Unser besonderes Interesse gilt den Nidoviren und der Frage, wie sie die größten bekannten RNA-Genome mit einer Länge von bis zu 64 Kilobasen entwickelt haben. Wir koordinieren die taxonomische Klassifizierung von Nidoviren im Rahmen des International Committee on Taxonomy of Viruses (ICTV).
PREDICTORix – ein Ansatz zur Quantifizierung des Risikos zoonotischer Übertragungen basierend auf Virus-Wirt Koevolution
PREDICTORix ist ein quantitativer, phylogeniebasierter Ansatz, um das Risiko zoonotischer Übertragungen über ein breites Spektrum von Virusfamilien hinweg abzuschätzen, ohne dass experimentelle Daten zu Übertragung oder Pathogenität erforderlich sind. Durch die Kombination globaler Sequenzressourcen (einschließlich >150.000 neu entdeckter RNA-Virussequenzen), der expliziten Modellierung evolutionärer Beziehungen zwischen Virus und Wirt sowie zweier Risikometriken (allgemeines und gezieltes Spillover-Risiko) generiert PREDICTORix quantitative Spillover-Werte, die als Leitfaden für Interventionen und mechanistische Folgemaßnahmen dienen können. Ursprünglich als Resilienzmaßnahme gegenüber menschlichen Pandemien entwickelt, lässt sich der Ansatz auf Tiere und andere Wirtsarten ausweiten und hilft dabei, potenzielle Kandidaten für „Pathogen X“ zu antizipieren.
Genetische Determinanten schwerer RSV-Infektionen
In enger Zusammenarbeit mit RESIST-Wissenschaftler:innen und Kliniker:innen der MHH untersuchen wir das komplexe Zusammenspiel genetischer Variationen im menschlichen Genom, um genetische Veränderungen zu identifizieren, die mit dem Verlauf oder der Schwere einer Infektion mit dem humanen Respiratorischen Synzytial-Virus (RSV) bei Kleinkindern assoziiert sind. Wir versuchen, sowohl monogene als auch polygene Faktoren zu identifizieren, die den Schweregrad der Erkrankung beeinflussen. Um diese und andere Fragen zu klären, haben unsere lokalen und internationalen Kooperationspartner die Exome von Kleinkindern mit schwerer RSV-Erkrankung sowie von Kontrollpersonen mit leichter RSV-Infektion sequenziert. Wir wenden Bioinformatik und genetische Assoziationsanalysen an, um wahrscheinliche kausale Varianten in immunbezogenen Genen zu identifizieren, die die Grundlage für experimentelle Folgeuntersuchungen bilden.
Prof. Chris Lauber
Wir zielen darauf ab, die stetig wachsende Zahl neu entdeckter Viren in evolutionäre Erkenntnisse zu überführen, die der Virologie und klinischen Forschung unmittelbar zugutekommen.

Chris Lauber schloss 2007 sein Bioinformatikstudium an der Universität Jena ab. Im Jahr 2012 promovierte er über Virusevolution und Bioinformatik in der Gruppe von Alexander Gorbalenya am Leiden University Medical Center, Leiden, Niederlande. Chris erhielt 2012 den NBIC Young Investigator Award für die beste in den Niederlanden verteidigte Doktorarbeit im Bereich Bioinformatik. Im Jahr 2013 wechselte er an die Technische Universität Dresden, wo er auf dem Gebiet der Virusentdeckung und OMICs-Datenanalyse arbeitete. Nach einem kurzen Aufenthalt als Datenwissenschaftler bei dem Biotech-Start-up Lipotype GmbH wurde er im Juni 2020 als Juniorprofessor (W1) an die Medizinische Hochschule Hannover im Rahmen des Exzellenzclusters RESIST berufen. Chris leitet die Forschungsgruppe „Computergestützte Virologie“ am TWINCORE. Seine Forschungsschwerpunkte sind Virusentdeckung und -evolution, Virustaxonomie, Pandemievorsorge, die Rolle von Viromen in Gesundheit und Krankheit, und Anfälligkeitsfaktoren für schwere Virusinfektionen.
Ausgewählte Publikationen
Haid S, Wetzke M, Todt D, Holwerda M, Dijkman R, Zillinger T, Jonigk D, Lange M, Kaderali L, Geffers R, Adhisantoso YG, Voges J, Ostermann J, Grethe C, Wiegmann B, Hansen G, Lauber C, Pietschmann T. TMEM259 alleles modulate respiratory syncytial virus infection and ER-stress-triggered apoptosis. [accepted in principal by Nature Communications]
Neuman BW, Smart A, Gilmer O, Smyth RP, Vaas J, Böker N, Samborskiy DV, Bartenschlager R, Seitz S, Gorbalenya AE, Caliskan N, Lauber C. Giant RNA genomes: Roles of host, translation elongation, genome architecture, and proteome in nidoviruses. Proc Natl Acad Sci U S A. 2025 Feb 18;122(7):e2413675122. doi: 10.1073/pnas.2413675122. Epub 2025 Feb 10. PMID: 39928875; PMCID: PMC11848433.
Lauber C, Zhang X, Vaas J, Klingler F, Mutz P, Dubin A, Pietschmann T, Roth O, Neuman BW, Gorbalenya AE, Bartenschlager R, Seitz S. Deep mining of the Sequence Read Archive reveals major genetic innovations in coronaviruses and other nidoviruses of aquatic vertebrates. PLoS Pathog. 2024 Apr 22;20(4):e1012163. doi: 10.1371/journal.ppat.1012163. PMID: 38648214; PMCID: PMC11065284.
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020 Apr;5(4):536-544. doi: 10.1038/s41564-020-0695-z. Epub 2020 Mar 2. PMID: 32123347; PMCID: PMC7095448.
Lauber C, Seitz S, Mattei S, Suh A, Beck J, Herstein J, Börold J, Salzburger W, Kaderali L, Briggs JAG, Bartenschlager R. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe. 2017 Sep 13;22(3):387-399.e6. doi: 10.1016/j.chom.2017.07.019. Epub 2017 Aug 31. PMID: 28867387; PMCID: PMC5604429.
Eine vollständige Liste der Publikationen finden Sie hier.
Newsroom
Sie haben Interesse an einer Bachelor- oder Masterarbeit? Wir freuen uns auf Ihre Anfrage!


