Computational Virology

Our Research
We are a computational research group that is interested in studying viruses and viral infections using sequence-based approaches. A common theme in our projects is the use of unprocessed data from public sequencing archives to generate and test scientific hypotheses in close collaboration with experimental virologists and clinicians. We develop and apply high-performance computing and deep learning workflows to analyze the vast volumes of data and metadata.
Data-Driven Virus Discovery
A major line of our research is the study of viral genetic diversity and virus evolution. We make use of the vast amount of public sequencing data, which we screen for the presence of viral genomes that have been sequenced as a by-product of sequencing the genome or transcriptome of the sampled organism. A main goal of our research is to comprehensively characterize the eukaryotic virome as well as the total virome of diseased and healthy humans. Moreover, we seek to discover unknown animal viruses that are closely related to pathogenic human viruses and may thus serve as experimental models. We have a special interest in nidoviruses and how they evolved the largest known RNA genomes, up to 64 kilobases. We are coordinating the taxonomic classification of nidoviruses as part of the International Committee on Taxonomy of Viruses (ICTV).
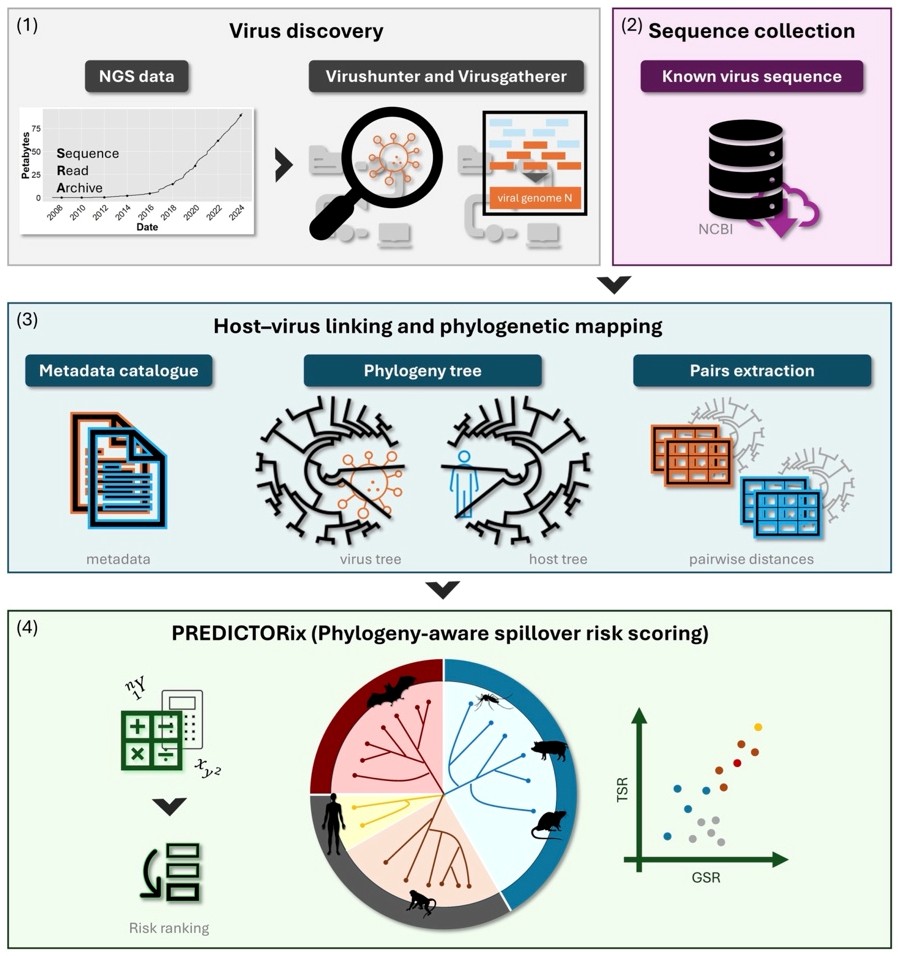
PREDICTORix – a virus-host co-evolutionary framework to quantify zoonotic spillover risk
PREDICTORix is a quantitative, phylogeny-aware framework that estimates zoonotic spillover risk across a broad range of virus families without requiring experimental data on transmission or pathogenicity. By combining global sequence resources (including >150,000 newly discovered RNA virus sequences), explicit modeling of virus–host evolutionary relationships, and dual risk metrics (General and Targeted Spillover Risk), PREDICTORix generates quantitative spillover scores to guide interventions and mechanistic follow-up. Originally developed for human pandemic resilience, the framework can be extended to animal and other host species and helps anticipate potential Pathogen X candidates.
Genetic determinants of severe RSV infection
In close collaboration with RESIST scientists and clinicians from MHH, we study the complex interplay of genetic variation in the human genome to identify genetic changes associated with the course or severity of infection with human respiratory syncytial virus (RSV) in young children. We seek to identify both monogenic and polygenic factors that influence disease severity. To address these and other questions, our local and international collaborators have sequenced the exomes of young children with severe RSV disease and of controls with mild RSV infection. We apply bioinformatics and genetic association analyses to identify likely causal variants in immunity-related genes that can form the basis for experimental follow-up.
Our Research
We are a computational research group that is interested in studying viruses and viral infections using sequence-based approaches. A common theme in our projects is the use of unprocessed data from public sequencing archives to generate and test scientific hypotheses in close collaboration with experimental virologists and clinicians. We develop and apply high-performance computing and deep learning workflows to analyze the vast volumes of data and metadata.
Data-Driven Virus Discovery
A major line of our research is the study of viral genetic diversity and virus evolution. We make use of the vast amount of public sequencing data, which we screen for the presence of viral genomes that have been sequenced as a by-product of sequencing the genome or transcriptome of the sampled organism. A main goal of our research is to comprehensively characterize the eukaryotic virome as well as the total virome of diseased and healthy humans. Moreover, we seek to discover unknown animal viruses that are closely related to pathogenic human viruses and may thus serve as experimental models. We have a special interest in nidoviruses and how they evolved the largest known RNA genomes, up to 64 kilobases. We are coordinating the taxonomic classification of nidoviruses as part of the International Committee on Taxonomy of Viruses (ICTV).
PREDICTORix – a virus-host co-evolutionary framework to quantify zoonotic spillover risk
PREDICTORix is a quantitative, phylogeny-aware framework that estimates zoonotic spillover risk across a broad range of virus families without requiring experimental data on transmission or pathogenicity. By combining global sequence resources (including >150,000 newly discovered RNA virus sequences), explicit modeling of virus–host evolutionary relationships, and dual risk metrics (General and Targeted Spillover Risk), PREDICTORix generates quantitative spillover scores to guide interventions and mechanistic follow-up. Originally developed for human pandemic resilience, the framework can be extended to animal and other host species and helps anticipate potential Pathogen X candidates.
Genetic determinants of severe RSV infection
In close collaboration with RESIST scientists and clinicians from MHH, we study the complex interplay of genetic variation in the human genome to identify genetic changes associated with the course or severity of infection with human respiratory syncytial virus (RSV) in young children. We seek to identify both monogenic and polygenic factors that influence disease severity. To address these and other questions, our local and international collaborators have sequenced the exomes of young children with severe RSV disease and of controls with mild RSV infection. We apply bioinformatics and genetic association analyses to identify likely causal variants in immunity-related genes that can form the basis for experimental follow-up.
Prof. Chris Lauber
We aim to channel the ongoing tsunami of virus discovery into evolutionary insights that are of direct benefit to virus and clinical research.

Chris Lauber completed his bioinformatics studies at the University of Jena in 2007. In 2012, he completed his PhD in the group of Alexander Gorbalenya at Leiden University Medical Center, Leiden, Netherlands, on the topic of virus evolution and bioinformatics. Chris received the NBIC Young Investigator award for the best PhD thesis in bioinformatics defended in the Netherlands in 2012. In 2013, he joined the University of Technology Dresden, working in the field of virus discovery and OMICs data analysis. After a short stay as a data scientist at the biotech start-up Lipotype GmbH, he was appointed junior professor (W1) at the Hannover Medical School in June 2020 as part of the RESIST cluster of excellence. Chris leads the research group “Computational Virology” at TWINCORE. His research focuses on virus discovery and evolution, virus taxonomy, pandemic preparedness, the role of viromes in health and disease, and susceptibility factors for severe viral infections.
Selected Publications
Haid S, Wetzke M, Todt D, Holwerda M, Dijkman R, Zillinger T, Jonigk D, Lange M, Kaderali L, Geffers R, Adhisantoso YG, Voges J, Ostermann J, Grethe C, Wiegmann B, Hansen G, Lauber C, Pietschmann T. TMEM259 alleles modulate respiratory syncytial virus infection and ER-stress-triggered apoptosis. [accepted in principal by Nature Communications]
Neuman BW, Smart A, Gilmer O, Smyth RP, Vaas J, Böker N, Samborskiy DV, Bartenschlager R, Seitz S, Gorbalenya AE, Caliskan N, Lauber C. Giant RNA genomes: Roles of host, translation elongation, genome architecture, and proteome in nidoviruses. Proc Natl Acad Sci U S A. 2025 Feb 18;122(7):e2413675122. doi: 10.1073/pnas.2413675122. Epub 2025 Feb 10. PMID: 39928875; PMCID: PMC11848433.
Lauber C, Zhang X, Vaas J, Klingler F, Mutz P, Dubin A, Pietschmann T, Roth O, Neuman BW, Gorbalenya AE, Bartenschlager R, Seitz S. Deep mining of the Sequence Read Archive reveals major genetic innovations in coronaviruses and other nidoviruses of aquatic vertebrates. PLoS Pathog. 2024 Apr 22;20(4):e1012163. doi: 10.1371/journal.ppat.1012163. PMID: 38648214; PMCID: PMC11065284.
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020 Apr;5(4):536-544. doi: 10.1038/s41564-020-0695-z. Epub 2020 Mar 2. PMID: 32123347; PMCID: PMC7095448.
Lauber C, Seitz S, Mattei S, Suh A, Beck J, Herstein J, Börold J, Salzburger W, Kaderali L, Briggs JAG, Bartenschlager R. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe. 2017 Sep 13;22(3):387-399.e6. doi: 10.1016/j.chom.2017.07.019. Epub 2017 Aug 31. PMID: 28867387; PMCID: PMC5604429.
A complete list of publications can be found here.
Newsroom
Are you interested in a bachelor or master thesis? We are looking forward to your request!


