Unsere Forschung
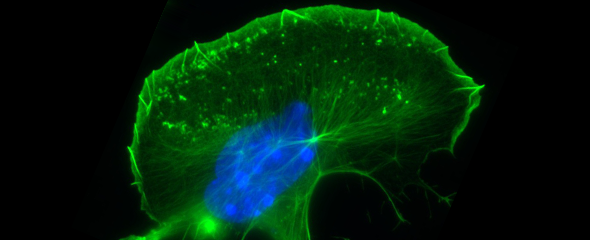
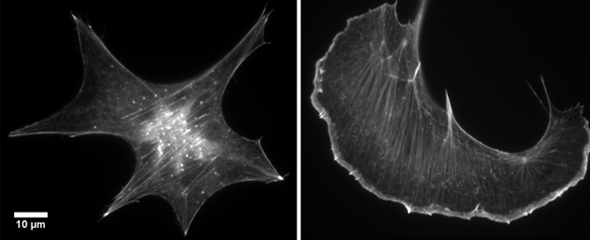
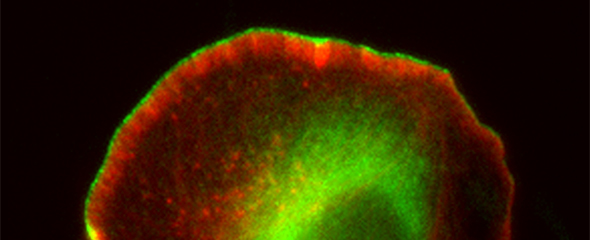
Unser Team beschäftigt sich vorwiegend mit den molekularen Mechanismen, die den Auf- und Umbau verschiedener zellulärer Aktinstrukturen steuern. Das Aktin-Zytoskelett bildet unterschiedlichste Strukturen mit unzähligen Funktionen aus. Diese reichen von hochorganisierten, kontraktilen Bündeln aus Aktin- und Myosinfilamenten, wie im Skelett- oder Herzmuskel zu finden, bis zu sehr transienten Aktinakkumulationen bei Endo- oder Phagozytose oder den hochdynamischen Zellausläufern migrierender Zellen, wie Lamellipodien und Filopodien.
Auch spezifische Varianten von Zell-Zell oder Zell-Substratkontakten werden meist direkt vom Aktinzytoskelett gebildet und reguliert, und haben entscheidende Funktionen für Zell- und Pathophysiologie wie beispielsweise die Invadopodien metastasierender Krebszellen.
Aktinabhängige Bewegungsprozesse sind für eine effektive Immunantwort und die Metastasenbildung von Tumorzellen gleichermaßen bedeutend, und stehen daher häufig im Zentrum anwendungsorientierter Grundlagenforschung.
Das Aktinzyoskelett wird aber auch vielfach von viralen oder bakteriellen Pathogenen missbraucht oder umfunktioniert, um in Wirtszellen einzudringen, sich dort zu verstecken oder zu verbreiten. Wir fokussieren uns daher hauptsächlich auf die molekularen Mechanismen der Aktinfilamentbildung und -reorganisation, vor allem an der Plasmamembran, und wie diese Prozesse durch bakterielle Pathogene benutzt werden.
Bei der Polymerisation von Aktinfilamenten sind zunächst Nukleation und Elongation entscheidend. Es werden mittlerweile drei Klassen von Nukleatoren unterschieden, und zwar der Arp2/3-Komplex und seine Aktivatoren, die Klasse der Formine und die Mitglieder der Spir/Cobl/Leiomodin-Gruppe.
Der Arp2/3-Komplex besteht aus sieben Untereinheiten, von denen zwei eine sehr hohe Homologie zu Aktin aufweisen (Arp2 und Arp3). Er ist in isolierter Form weitgehend inaktiv und muss daher von zusätzlichen Faktoren aktiviert werden. Dies wird durch eine Familie von Proteinen bewerkstelligt, deren Namensgeber in einer seltenen Immunschwäche mutiert ist, dem Wiskott-Aldrich-Syndrom. Das zugehörige, im hämatopoetischen System exprimierte Genprodukt heißt daher WAS-Protein, oder kurz WASP. Im Säuger gibt es neben WASP, dem ubiquitären N-WASP (neural WASP da stark im Nervensystem angereichert) und drei WAVE-Isoformen (WAVE 1, 2, 3) noch die erst in jüngerer Zeit identifizierten Mitglieder WASH, WHAMM und JMY. Diese acht Aktivatoren des Arp2/3-Komplexes werden auch als Typ I Nukleationspromotionsfaktoren bezeichnet (NPF).
WASP, N-WASP und WAVE-Proteine sind direkte oder indirekte Effektoren der kleinen GTPasen der Rho (Ras homology) – Familie, deren bekannteste Mitglieder Rac1, RhoA und Cdc42 die Ausbildung zellulärer Aktinstrukturen stimulieren. Man geht heute davon aus, dass beispielsweise die durch die kleine Rho-GTPase Rac getriebene Ausbildung von Lamellipodien durch die direkte Verknüpfung mit WAVE und der damit einhergehenden Aktivierung des Arp2/3-Komplexes bewerkstelligt wird. Obwohl Rac/WAVE- und Arp2/3-Komplex-abhängige Aktinpolymerisation essentiell für die Ausbildung lamellipodialer Aktinnetzwerke sind, haben auch weitere durch Rho-GTPasen gesteuerte Signalwege einen Einfluss. So konnten wir beispielsweise zeigen, dass die kleine GTPase Cdc42 die Ausbildung dieser Strukturen durch die Aktivierung von FMNL2- und -3, Mitglieder der Formin-Familie beeinflusst.
Eine weitere Gruppe von Bindeproteinen des Arp2/3-Komplexes sind die Typ II NPFs, zu denen im Säuger das ubiquitäre Src-Kinase-Substrat Cortactin und das hämatopoetische HS1 gezählt werden. Cortactin und HS1 ko-lokalisieren häufig mit dem Arp2/3-Komplex an hochdynamischen Aktinstrukturen, ihre genaue Funktion in diesen Strukturen ist aber noch weitgehend unverstanden. Wir bearbeiten in unserer Gruppe unterschiedliche Mausmodelle mit defekter Cortactin und/oder HS1-Expression, und untersuchen Funktionen in unterschiedlichsten Geweben und Krankheitsprozessen.
In jüngerer Vergangenheit haben wir damit begonnen, die oben genannten Signalwege, beispielsweise stimuliert durch die kleine GTPase Rac und ihrer verwandten GTPasen wie Cdc42 oder auch RhoG mittels CRISPR/Cas9 molekular im Detail zu analysieren. Das selektive Entfernen einzelnen Glieder dieser Signalketten wird dabei zunehmend durch Rekonstitution dieser Signalwege mit Mutanten unterschiedlichster Aktivität ersetzt, was völlig neue Dimensionen unseres Verständnisses der molekularen Mechanismen dieser Signalwege erlaubt. Wir postulieren, dass solch systematische Analysen kombiniert mit mathematischer Modellierung der Veränderungen der biochemischen Aktivitäten bei unterschiedlichen experimentellen Bedingungen die Entwicklung völlig neuer Modelle des Funktionierens spezifischer Aktinstrukturen bei Motilitäts- und Pathogen-Wirtszellinteraktionsprozessen erlauben wird.
Unsere Arbeiten profitieren von zahlreichen, sehr fruchtbaren nationalen und internationalen Kooperationen wie beispielsweise mit den Laboren von:
- Jan Faix (Hannover),
- Cord Brakebusch (Kopenhagen, Dänemark),
- Michael Sixt (Klosterneuburg, Österreich),
- Michael Schnoor (Mexico Stadt) oder
- Grégory Giannone (Bordeaux, Frankreich).
In unterschiedlichen, beispielsweise von der Deutschen Forschungsgemeinschaft (DFG) finanzierten Projekten werden molekularen Manipulationen in Zellen mit hochauflösender Mikroskopie und biophysikalischen Messungen sowie mathematischer Modellierung (Martin Falcke, Berlin) verbunden. An HZI und TU Braunschweig werden zahlreiche Kooperationen neu etabliert oder vorangetrieben, beispielsweise mit den HZI-Abteilungen Strukturbiologie (Wulf Blankenfeldt), Zellbiologie (Theresia Stradal) oder den AGs ZEIM (Manfred Rohde) und CPRO (Lothar Jänsch), und an der TU mit Martin Korte und Kristin Michaelsen-Preusse (Zoologisches Institut) oder auch Peter Jomo Walla (Institut für Physikalische und Theoretische Chemie).
Unsere Forschung
Unser Team beschäftigt sich vorwiegend mit den molekularen Mechanismen, die den Auf- und Umbau verschiedener zellulärer Aktinstrukturen steuern. Das Aktin-Zytoskelett bildet unterschiedlichste Strukturen mit unzähligen Funktionen aus. Diese reichen von hochorganisierten, kontraktilen Bündeln aus Aktin- und Myosinfilamenten, wie im Skelett- oder Herzmuskel zu finden, bis zu sehr transienten Aktinakkumulationen bei Endo- oder Phagozytose oder den hochdynamischen Zellausläufern migrierender Zellen, wie Lamellipodien und Filopodien.
Auch spezifische Varianten von Zell-Zell oder Zell-Substratkontakten werden meist direkt vom Aktinzytoskelett gebildet und reguliert, und haben entscheidende Funktionen für Zell- und Pathophysiologie wie beispielsweise die Invadopodien metastasierender Krebszellen.
Aktinabhängige Bewegungsprozesse sind für eine effektive Immunantwort und die Metastasenbildung von Tumorzellen gleichermaßen bedeutend, und stehen daher häufig im Zentrum anwendungsorientierter Grundlagenforschung.
Das Aktinzyoskelett wird aber auch vielfach von viralen oder bakteriellen Pathogenen missbraucht oder umfunktioniert, um in Wirtszellen einzudringen, sich dort zu verstecken oder zu verbreiten. Wir fokussieren uns daher hauptsächlich auf die molekularen Mechanismen der Aktinfilamentbildung und -reorganisation, vor allem an der Plasmamembran, und wie diese Prozesse durch bakterielle Pathogene benutzt werden.
Bei der Polymerisation von Aktinfilamenten sind zunächst Nukleation und Elongation entscheidend. Es werden mittlerweile drei Klassen von Nukleatoren unterschieden, und zwar der Arp2/3-Komplex und seine Aktivatoren, die Klasse der Formine und die Mitglieder der Spir/Cobl/Leiomodin-Gruppe.
Der Arp2/3-Komplex besteht aus sieben Untereinheiten, von denen zwei eine sehr hohe Homologie zu Aktin aufweisen (Arp2 und Arp3). Er ist in isolierter Form weitgehend inaktiv und muss daher von zusätzlichen Faktoren aktiviert werden. Dies wird durch eine Familie von Proteinen bewerkstelligt, deren Namensgeber in einer seltenen Immunschwäche mutiert ist, dem Wiskott-Aldrich-Syndrom. Das zugehörige, im hämatopoetischen System exprimierte Genprodukt heißt daher WAS-Protein, oder kurz WASP. Im Säuger gibt es neben WASP, dem ubiquitären N-WASP (neural WASP da stark im Nervensystem angereichert) und drei WAVE-Isoformen (WAVE 1, 2, 3) noch die erst in jüngerer Zeit identifizierten Mitglieder WASH, WHAMM und JMY. Diese acht Aktivatoren des Arp2/3-Komplexes werden auch als Typ I Nukleationspromotionsfaktoren bezeichnet (NPF).
WASP, N-WASP und WAVE-Proteine sind direkte oder indirekte Effektoren der kleinen GTPasen der Rho (Ras homology) – Familie, deren bekannteste Mitglieder Rac1, RhoA und Cdc42 die Ausbildung zellulärer Aktinstrukturen stimulieren. Man geht heute davon aus, dass beispielsweise die durch die kleine Rho-GTPase Rac getriebene Ausbildung von Lamellipodien durch die direkte Verknüpfung mit WAVE und der damit einhergehenden Aktivierung des Arp2/3-Komplexes bewerkstelligt wird. Obwohl Rac/WAVE- und Arp2/3-Komplex-abhängige Aktinpolymerisation essentiell für die Ausbildung lamellipodialer Aktinnetzwerke sind, haben auch weitere durch Rho-GTPasen gesteuerte Signalwege einen Einfluss. So konnten wir beispielsweise zeigen, dass die kleine GTPase Cdc42 die Ausbildung dieser Strukturen durch die Aktivierung von FMNL2- und -3, Mitglieder der Formin-Familie beeinflusst.
Eine weitere Gruppe von Bindeproteinen des Arp2/3-Komplexes sind die Typ II NPFs, zu denen im Säuger das ubiquitäre Src-Kinase-Substrat Cortactin und das hämatopoetische HS1 gezählt werden. Cortactin und HS1 ko-lokalisieren häufig mit dem Arp2/3-Komplex an hochdynamischen Aktinstrukturen, ihre genaue Funktion in diesen Strukturen ist aber noch weitgehend unverstanden. Wir bearbeiten in unserer Gruppe unterschiedliche Mausmodelle mit defekter Cortactin und/oder HS1-Expression, und untersuchen Funktionen in unterschiedlichsten Geweben und Krankheitsprozessen.
In jüngerer Vergangenheit haben wir damit begonnen, die oben genannten Signalwege, beispielsweise stimuliert durch die kleine GTPase Rac und ihrer verwandten GTPasen wie Cdc42 oder auch RhoG mittels CRISPR/Cas9 molekular im Detail zu analysieren. Das selektive Entfernen einzelnen Glieder dieser Signalketten wird dabei zunehmend durch Rekonstitution dieser Signalwege mit Mutanten unterschiedlichster Aktivität ersetzt, was völlig neue Dimensionen unseres Verständnisses der molekularen Mechanismen dieser Signalwege erlaubt. Wir postulieren, dass solch systematische Analysen kombiniert mit mathematischer Modellierung der Veränderungen der biochemischen Aktivitäten bei unterschiedlichen experimentellen Bedingungen die Entwicklung völlig neuer Modelle des Funktionierens spezifischer Aktinstrukturen bei Motilitäts- und Pathogen-Wirtszellinteraktionsprozessen erlauben wird.
Unsere Arbeiten profitieren von zahlreichen, sehr fruchtbaren nationalen und internationalen Kooperationen wie beispielsweise mit den Laboren von:
- Jan Faix (Hannover),
- Cord Brakebusch (Kopenhagen, Dänemark),
- Michael Sixt (Klosterneuburg, Österreich),
- Michael Schnoor (Mexico Stadt) oder
- Grégory Giannone (Bordeaux, Frankreich).
In unterschiedlichen, beispielsweise von der Deutschen Forschungsgemeinschaft (DFG) finanzierten Projekten werden molekularen Manipulationen in Zellen mit hochauflösender Mikroskopie und biophysikalischen Messungen sowie mathematischer Modellierung (Martin Falcke, Berlin) verbunden. An HZI und TU Braunschweig werden zahlreiche Kooperationen neu etabliert oder vorangetrieben, beispielsweise mit den HZI-Abteilungen Strukturbiologie (Wulf Blankenfeldt), Zellbiologie (Theresia Stradal) oder den AGs ZEIM (Manfred Rohde) und CPRO (Lothar Jänsch), und an der TU mit Martin Korte und Kristin Michaelsen-Preusse (Zoologisches Institut) oder auch Peter Jomo Walla (Institut für Physikalische und Theoretische Chemie).
Prof. Dr. Klemens Rottner

Klemens Rottner absolvierte sein Studium der Biologie/Zoologie an der Paris-Lodron-Universität in Salzburg und promovierte anschließend im Bereich Zellbiologie/Immunologie. Im Jahr 2000 entschied er sich für einen Forschungsaufenthalt in Braunschweig und arbeitete dort als Postdoc in der Gesellschaft für Biotechnologische Forschung. Im Anschluss übernahm er 2004 die Leitung der Forschungsgruppe Zytoskelett Dynamik.
2010 folgte er dem Ruf der Rheinischen Friedrich-Wilhelms-Universität in Bonn und übernahm eine Professur am Institut für Genetik. Im Jahr 2014 kehrte er nach Braunschweig zurück. Seither ist er als Professor am Institut für Zoologie der Technischen Universität tätig und leitet zudem die Arbeitsgruppe Molekulare Zellbiologie am Helmholtz-Zentrum für Infektionsforschung.
Team






Ausgewählte Publikationen
Rottner, K., Stradal, T.E.B., Chen, B. (2021) WAVE regulatory complex. Curr Biol, 31(10):R512-R517. DOI: 10.1016/j.cub.2021.01.086.
Dimchev, V., Lahmann, I., Koestler, S.A., Kage, F., Dimchev, G., Steffen, A., Stradal, T.E.B., Vauti, F., Arnold, H.H., Rottner, K. (2021) Induced Arp2/3 complex depletion increases FMNL2/3 formin expression and filopodia formation. Front Cell Dev Biol, 9:634708. DOI: 10.3389/fcell.2021.634708.
Dimchev, G., Amiri, B., Humphries, A.C., Schaks, M., Dimchev, V., Stradal, T.E., Faix, J., Krause, M., Way, M., Falcke, M., Rottner, K. (2020) Lamellipodin tunes cell migration by stabilizing protrusions and promoting adhesion formation. J Cell Sci, 133(7):jcs239020.
Schaks, M., Singh, S.P., Kage, F., Thomason, P., Klünemann, T., Steffen, A., Blankenfeldt, W., Stradal, T.E., Insall, R., Rottner, K. (2018) Distinct interaction sites of Rac GTPase with WAVE regulatory complex have non-redundant functions in vivo. Curr Biol, 28(22):3674-3684. doi: 10.1016/j.cub.2018.10.002.
Kage, F., Winterhoff, M., Dimchev, V., Mueller, J., Thalheim, T., Freise, A., Brühmann, S., Kollasser, J., Block, J., Dimchev, G., Geyer, M., Schnittler, H.-J., Brakebusch, C., Stradal, T.E., Carlier, M.-F., Sixt, M., Käs, J., Faix, J., Rottner, K. (2017) FMNL formins boost lamellipodial force generation. Nat Commun, 8:14832.
Publikationen
Aktuelle Projekte
Dissection of formin functions in cell edge protrusion and migration
Projektpartner:
- Prof. Dr. Jan Faix (Hannover)
- Prof. Dr. Michael Sixt (Klosterneuburg, Austria)
- Dr. Florian Schur (Klosterneuburg, Austria)
Geldgeber/Förderer:
- DFG - Deutsche Forschungsgemeinschaft
Project P4: Lamellipodial protein complexes driving Arp2/3-dependent branching (PROCOMPAS)
Geldgeber/Förderer:
- DFG - Deutsche Forschungsgemeinschaft
Newsroom